

What is a metagenomic analysis?
Metagenomic analysis involves analysis of genetic material collected directly from environmental or clinical samples, or any other sample type containing a microbiome, i.e., more than one microbial species. For instance, such samples could be: the human intestine, oral cavity, wastewater, soil samples, etc. This type of study aims to understand the structure, function, and diversity of microbial communities present in these samples by examining their collective genetic material. Traditional microbiology methods enable detection only of a subset of microbes in a sample (only those organisms that can be cultured under laboratory conditions). Metagenomics (the use of NGS, next-generation sequencing, on complex microbial samples) overcomes this limitation by directly extracting and sequencing DNA or RNA from environmental samples, providing a snapshot of the genetic content of the entire microbial community.
Why is VeriFi® Library Amplification Mix ideal for metagenomic analyses?
VeriFi® Library Amplification Mix offered 2% more deduplicated reads (or 2% fewer duplicated reads) in three different microbial genomes, compared to other dedicated library amplification mixes KAPA HiFi HotStart Library Amplification Kit, NEBNext® Ultra™ II Q5® Master Mix, and Takara SeqAmp™ DNA Polymerase, in a blind experiment conducted by external testers (Figure 1). Depending on the sequencing platform used this could translate to 80,000-25 million more unique reads per dataset.
Additionally, the mix has demonstrably lower GC-bias than any competitor mix tested (>5 other mixes, Figure 2).
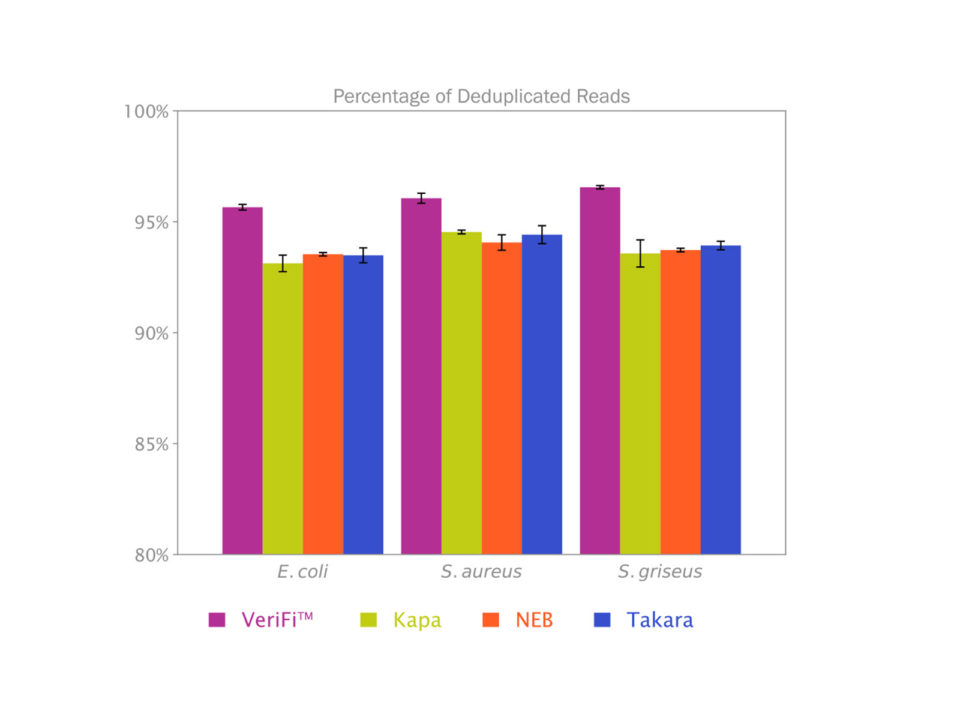
Figure 1. The number of uniquely mapped reads for three microbial genomes with different average GC content (E. coli ~50% GC, S. aureus ~30% GC, and S. griseus ~70% GC) shown as a percentage of total reads in four sequencing datasets. Datasets were generated using Illumina® sequencing in a blind experiment where all three genome libraries were amplified with different proofreading polymerases, VeriFi® Library Amplification Mix (purple), KAPA HiFi HotStart Library Amplification Kit (green), NEBNext® Ultra™ II Q5® Master Mix (orange), and Takara SeqAmp™ DNA Polymerase (blue).
NGS library amplification with VeriFi® Library Amplification Mix leads to a higher number of unique reads per dataset after read deduplication compared to leading competitors.
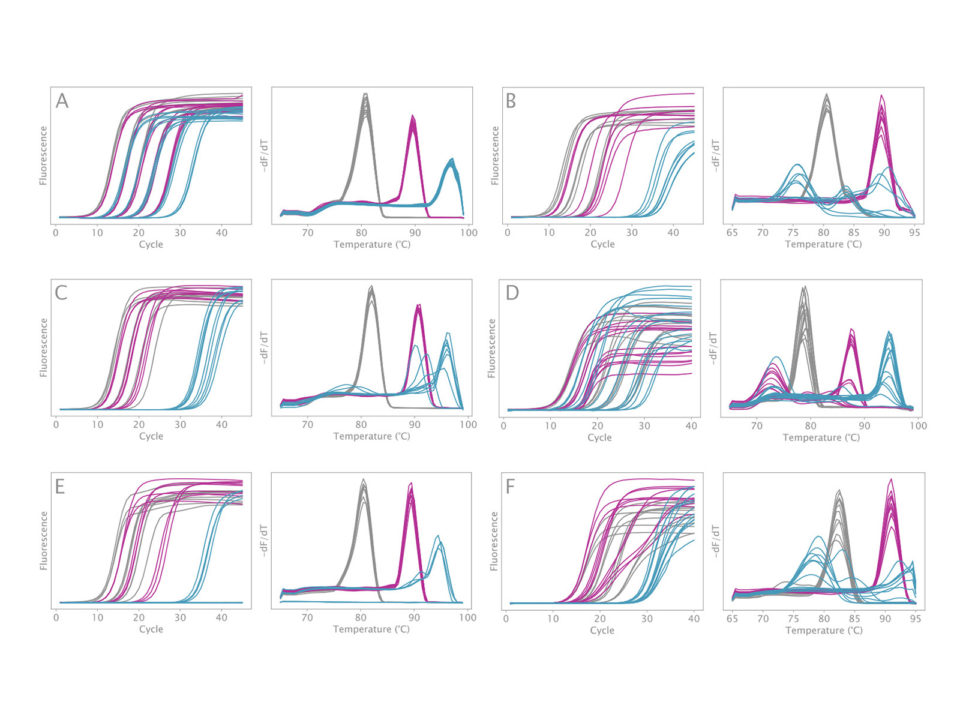
Figure 2. Amplification of synthetic 1 kb sequences with different GC contents. Grey: 30% GC; Purple: 50% GC; Blue: 70% GC. Amplification curves are shown on the left panel and melt peaks on the right panel. Serial dilutions of each template were used in a reaction volume of 20 µL. Mixes tested are A) VeriFi® Library Amplification Mix, B) KAPA HiFi HotStart Library Amplification Kit, C) NEBNext® Ultra™ II Q5® Master Mix, D) RepliQa HiFi PCR Master Mix (Quantabio), E) Platinum™ SuperFi II DNA Polymerase (Thermo), and F) PrimeStar GXL DNA Polymerase (Takara). Each mix was run under cycling conditions recommended by the manufacturer.
VeriFi® Library Amplification Mix consistently amplifies templates across a broad range of GC content and over a wide range of concentrations with much less bias than competitors.
Why it is important to reduce read duplication in NGS datasets for metagenomic analysis1:
- Accurate representation of microbial diversity: Metagenomic analyses aim to capture the diversity of microbial organisms present in a sample. Duplicated reads arise when the same DNA fragment is sequenced multiple times, leading to an overestimation of the abundance of certain organisms and a distortion of the overall microbial composition. By minimizing duplicates, researchers can obtain a more accurate representation of the true microbial diversity within the sample.
- Reliable abundance estimation: Duplicated reads can skew abundance estimates of microbial taxa. If a particular DNA fragment is overrepresented due to duplication, it will artificially inflate the abundance of the corresponding organism or gene. This can lead to erroneous conclusions about the relative abundance and importance of different taxa or functional genes in the microbial community. By reducing duplicated reads, researchers can obtain more reliable estimates of microbial abundance.
- Enhanced statistical power: Metagenomic analyses often involve statistical comparisons between different samples or conditions. High levels of duplicated reads can introduce bias and reduce the statistical power of these analyses. By minimizing duplicates, researchers can improve the accuracy of statistical tests and increase their ability to detect meaningful differences between samples.
- Cost-effectiveness: Sequencing and analysing metagenomic data can be expensive, both in terms of time and resources. Duplicated reads consume unnecessary sequencing capacity and computational resources without adding valuable information. Reducing duplication, can optimise sequencing efforts, leading to cost savings and more efficient data analysis.
Why reducing GC-bias in metagenomic analyses is important2:
- Accurate representation of microbial diversity: GC-bias refers to the preferential amplification or sequencing of DNA fragments with specific GC content. If there is a significant bias towards certain GC content ranges, it can result in an inaccurate representation of the microbial diversity in the sample. Some microbial taxa may be overrepresented, while others may be underrepresented, leading to a distortion of the relative abundances and composition of the microbial community. Minimizing GC-bias helps ensure a more accurate and unbiased representation of microbial diversity.
- Improved taxonomic and functional profiling: GC-bias can impact the accuracy of taxonomic and functional profiling of metagenomic data. If certain GC content ranges are overrepresented, it can lead to false identifications and misclassification of microbial taxa. Similarly, functional genes associated with specific GC content ranges may be disproportionately represented, leading to biased functional profiling. By reducing GC-bias, researchers can improve the reliability and accuracy of taxonomic and functional assignments, enabling more robust biological interpretations.
- Enhanced comparative analyses: Metagenomic analyses often involve comparing multiple samples or conditions. GC-bias can introduce systematic differences in read coverage and abundance estimates, making it challenging to perform accurate and meaningful comparisons. Minimizing GC-bias reduces the variability introduced by biased amplification and sequencing, improving the comparability of different samples and enabling more reliable statistical analyses.
- Improved downstream analysis: GC-bias can have implications for downstream analyses, such as assembly, gene prediction, and pathway reconstruction. Biased coverage and incorrect abundance estimations resulting from GC-bias can affect the accuracy and reliability of these analyses. Reduced GC-bias can therefore improve the quality of downstream analysis outputs and increase confidence in the biological insights derived from metagenomic data.
VeriFi® Library Amplification Mix facilitates better quality library preparation by reducing GC-bias and enabling more efficient multiplexing. These, in turn, ensure metagenomic analyses provide accurate and meaningful insights into microbial communities.
To find more information, request a sample to test in your workflow, or get a quotation, visit our product webpage.
References
- Nayfach S, Pollard KS. Toward Accurate and Quantitative Comparative Metagenomics. Cell. 2016 Aug 25;166(5):1103-1116. doi: 10.1016/j.cell.2016.08.007. PMID: 27565341; PMCID: PMC5080976.
- Patrick D B, Nielsen T K, Kot W, Aggerholm A, Gilbert M T P, Puetz L, Rasmussen M, Zervas A, Hansen L H, GC bias affects genomic and metagenomic reconstructions, underrepresenting GC-poor organisms, GigaScience, Volume 9, Issue 2, February 2020, giaa008, doi: 10.1093/gigascience/giaa008